相信很多学CS的同学之前都没听说过“蛋白质结构预测”这个问题,直到2018年12月初,一则劲爆消息瞬间引爆了CSer的朋友圈,那就是Google Deepmind团队开发的AlphaFold一举拿下当年的CASP比赛冠军,而且远远甩开了第二名。我当时就转载过类似的公众号文章,大家可以阅读并想象当时朋友圈的欢呼声:阿尔法狗再下一城 | 蛋白结构预测AlphaFold大胜传统人类模型。
当时,很多同学也转载过类似的文章,但其实很少有人真正明白“蛋白质结构预测”这个问题是什么,它的难度有多大,CASP是个什么比赛,以及AlphaFold的内部原理是什么。当然,对于这一连串的问题,我当时也是懵逼的。不过自己好歹也是个跟蛋白质有关的PhD,如此热点事件,自然是要关注的。不过之后一直没时间,直到今年相关顶级文章再次爆出,我就借着准备文献讲评的机会了解了相关的知识,在这里跟大家分享一下。

蛋白质结构分为四级,分别是一级结构、二级结构、三级结构和四级结构,下面分别描述。
一级结构
蛋白质的一级结构可以理解为一条线性的字符串,比如MSFIKTFSGKHFYYDKINKDDIVINDIAVSLSNICR。其基本组成单元是一个个的氨基酸,即一个个的字母。氨基酸有单字母表示和三字母表示,为了简洁,本文使用单字母表示,下图的例子是三字母表示。常见的氨基酸只有20种,所以一级结构的字符串通常只包含20种字母,不包含的6种字母是BJOUXZ。

20种氨基酸的结构符合一个通式,如下图所示,中间的碳原子称为Cα碳原子,表示它处在α位;左边连了一个氨基-NH2,称为N端;右边连了一个羧基-COOH,称为C端。20种不同氨基酸的差别就在于Cα上连接的侧链基团R,具体的差别网上一搜就能查到。

20种氨基酸连接的方式为脱水缩合,即一个氨基酸的羧基-COOH和另一个氨基酸的氨基-NH2反应,丢掉一个H2O,形成一个肽键-CO-NH-,如下图所示。丢掉了羧基和氨基的氨基酸被称为氨基酸残基,这个名词很形象,氨基酸缺胳膊少腿,所以变成了“残”基。

二级结构
二级结构就是在一级结构的字符串的基础上,肽链怎样进行盘旋、折叠等变换,形成一种局部的三维结构,这种局部的三维结构通常由氢键支撑。常见的二级结构有α螺旋和β折叠,如下图所示。其中α螺旋的每个残基的-NH的H和临近的第4个残基的-CO的O形成氢键,由此支撑α螺旋的结构稳定性,如下图的箭头所指虚线。β折叠则是两条肽链,平行排列,对应残基的-NH的H和-CO的O形成氢键,由此形成两股β折叠的结构,多股β折叠形成类似手风琴的样子。β折叠分为平行和反平行排列,我们前面介绍到肽段分为N端和C端,如果形成β折叠的两股链都是从N到C(或从C到N),则称为平行排列,否则是反平行排列。每股β折叠都有一个大箭头表示其方向。


细分的话,蛋白质的二级结构总共有8种,包括转角、无规则卷曲等。目前常采用DSSP的分类方法,有些文献会把8种结构粗分为α螺旋、β折叠和转角这三种结构。
由上图可知,蛋白质的二级结构极大的决定了其三级结构(下面介绍),所以有很多工作是研究怎样准确预测蛋白质的二级结构的,即预测每个氨基酸残基处于哪一种二级结构中。形式化表示就是,对于一个蛋白质一级结构字符串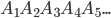 ,输出
,输出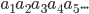 ,其中
,其中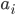 ∈{α螺旋,β折叠,转角}。所以,蛋白质的二级结构是一个端到端的问题,很像机器翻译,目前很多文章都会用深度学习NLP的方法来预测蛋白质的二级结构。
∈{α螺旋,β折叠,转角}。所以,蛋白质的二级结构是一个端到端的问题,很像机器翻译,目前很多文章都会用深度学习NLP的方法来预测蛋白质的二级结构。
三级结构
简单理解,三级结构就是把多个二级结构拼接到一起,折叠成一个完整的蛋白质三维结构,如下图所示。维持蛋白质三级结构的力比较多样,除了氢键之外,还有二硫键、金属键等。


四级结构
简单理解,四级结构就是多个三级结构分子组合成一个复合物,就是四级结构。

对于CSer来说,由于四级结构仅仅是多个三级结构组合到一起,我们常说的蛋白质三维结构预测问题,通常是指预测蛋白质的三级结构。问题是,构成蛋白质链的原子非常多,我们怎样形式化描述一条蛋白质的三维结构呢?这还要从最原始的一级结构说起。
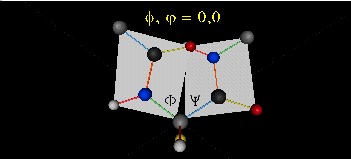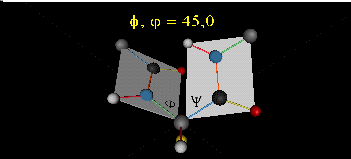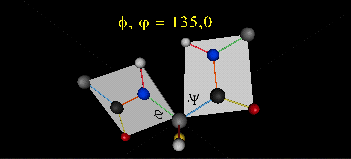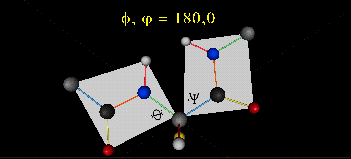
前面提到,两个氨基酸通过脱水缩合的方式形成肽键从而连接到一起形成一级结构(本文图四),肽键虽然是单键,但它具有类似双键的特点,即难以旋转(比如羧基中的-C=O键就是双键,无法旋转)。所以,由肽键及周围的6个原子形成了一个固定的肽键平面,这6个原子分别是-C-CO-NH-C-,如下图所示,箭头所指的红色键就是肽键,它周围画出了一个平面,就是肽键平面。


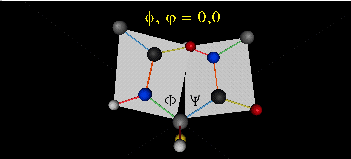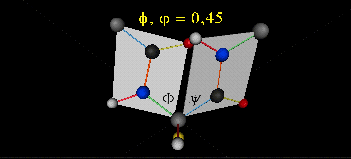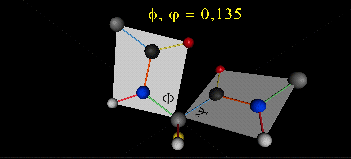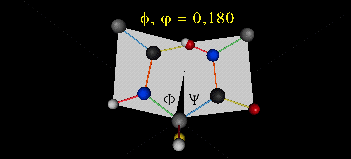
肽键平面的存在极大的简化了蛋白质结构,可以认为这6个原子的相对位置是固定的了!另一方面,跟这个平面相连的左右两个C原子的两个键是单键,所以他们可以旋转,旋转的角度称为扭转角ϕ和ψ,为了更直观的感受肽链的肽键平面和两个扭转角,可以看下面的动画:
事实上,扭转角ϕ和ψ并不是在360°范围内随机均匀分布的,1963年就有科学家统计过扭转角ϕ和ψ的分布,他们发现稳定的蛋白质结构的ϕ和ψ通常只分布在一小部分区域,如下图的拉氏图所示,这些区域正好对应了常见的α螺旋和β折叠的结构。

最后,我们还需要介绍一个角度,那就是ω。前面提到,虽然肽键具有双键的特点,难以旋转,但它在少数情况下还是可以旋转的。假设通常情况下,肽键的角度定义为ω=0°,如下图所示,红色的键即为肽键,这种结构的好处是它能让形成肽键的两个残基的侧链R(图中黑色基团)离得尽量的远,这样能保持比较稳定的结构。如果肽键旋转为ω=180°,变为下图的样子,则两个侧链R很靠近,就产生位阻效应,就不稳定,所以这种情况比较少见。但不管怎么说,肽键的扭转角ω也是一个变量因素。

综上所述,对于一条肽链,如果知道每个残基的三个扭转角ϕ、ψ和ω,则可以重构出肽链的主干部分的三维结构,这就像将极坐标转换为直角坐标一样容易。需要提醒的是,本文提到的蛋白质三维结构预测问题,对蛋白质的结构进行了简化,包括:1. 仅预测蛋白质或肽链的主干结构,不考虑侧链R的结构;2. 假设肽链主干中每个键的长度是固定的;3. 不考虑键的角度,比如对于上图的肽键,仅考虑肽键绕肽键轴本身的旋转,不考虑肽键绕着某一端原子的旋转,比如固定左边的蓝色小球,肽键和右边的红色小球旋转出平面了。 下图的肽键平面,详细的标识出了各个相对固定的值。

所以,对于CSer来说,蛋白质的三维结构预测问题,就可以看成一个端到端的学习问题,输入是一个字符串,输出是每个字符(残基)对应的三个扭转角ϕ、ψ和ω,问题看起来非常的简洁漂亮。而且,这个问题和NLP中的序列标注、机器翻译等问题很像,所以很多NLP的技术可以用来预测蛋白质的三维结构。下图的插画就是最近发表在Cell Sytems上的一篇用LSTM预测蛋白质三维结构的文章,我会在下一篇博客中和大家分享这篇文章。

有关“蛋白质结构预测”本身的最后一个问题是,为什么能仅仅通过一级结构的序列信息,预测得到其三级结构呢?也就是说蛋白质结构预测这个问题是否可解,如果蛋白质的三级结构还由其他因素决定,那么即使Deeplearning玩出花了,在生物上也是不可行的。所以,每遇到一个新问题,都要自问一下,这个问题从原理上是否可解。对于“蛋白质结构预测”这个问题,最开始也有人进行了类似的自问,得到的答案是可行的:
1965年,安芬森(Anfinsen)基于还原变性的牛胰RNase在不需其他任何物质帮助下,仅通过去除变性剂和还原剂就使其恢复天然结构的实验结果,提出了“多肽链的氨基酸序列包含了形成其热力学上稳定的天然构象所必需的全部信息”的“自组装学说”,随后这个学说又得到一些补充。这些学说表明:氨基酸序列确定其空间构象,从而为蛋白质结构预测提供了可行性。
http://chinaxiv.org/user/download.htm?id=6478
提到蛋白质三级结构预测,不得不提的是CASP这个比赛。CASP的全称是The Critical Assessment of protein Structure Prediction (CASP),即蛋白质结构预测的关键评估,被誉为蛋白质结构预测的奥林匹克竞赛。CASP从1994年开始举办,每两年一届,最近的一届是2018年的CASP13。
每一届CASP比赛,都会提供大约100条未知结构的蛋白质序列,让所有参赛者进行结构预测,比赛结束之后,主办方会通过生化方法测定这些蛋白质的三维结构,然后和参赛者预测的结果进行比对,然后给出预测得分。提供的蛋白质序列分为两类:一类序列和PDB数据库中已有结构的序列有相似性,由此可以基于模板预测,准确度比较高,这类算法称为Template-Based Modeling;另一类序列和PDB库已知结构的序列相似度很低,可以认为是全新的蛋白质,因为无法利用已有模板信息,需要进行从头测序(De novo或ab initio或Free Modeling),目前的准确率比较低。参赛选手也分为两组,一组是servers only,即仅允许算法参赛,给定3天的时间;另一组是human and servers,即允许人和算法合作,共同预测蛋白质结构,给定3周的时间。
CASP同时提供多种比赛项目,比如常规的结构预测(Regular targets)、数据辅助预测(Data-Assisted targets)和蛋白质接触面预测(Contact predictions)等,其中数据辅助预测中提供了核磁数据(NMR)、交联数据(XLMS)等,对的,交联数据就是我目前研究的pLink处理的数据。

AlphaFold参加了CASP13的humans and servers组,第一次参赛就一鸣惊人,获得冠军并甩开第二名好多。

在蛋白质结构预测领域,活跃着很多华人学者,比如密西根大学张阳团队在servers only组获得七连冠CASP7~CASP13,14年霸主地位! (I-TASSER (as Zhang-Server) and QUARK from Zhang Lab)。大家可以围观一下张老师实验室的机房以及张老师在清华的一个访谈。另外,芝加哥大学的许锦波团队开发的RaptorX在每届的CASP上也取得了不错的成绩。国内方面,中科院计算所的卜东波老师和上海交大的沈红斌老师也有相关的研究。

OK,以上就是有关“蛋白质结构预测”这个问题的入门介绍,由于本人并非研究这个领域,理解有误的地方还请大家指出。最后,在入门这个领域时,我查了很多资料,上面的图片也都来自网络并注明了出处,下面汇总列出供大家参考:
蛋白质结构基础知识:
- 维基百科:https://en.wikipedia.org/wiki/Protein_structure
- 俄勒冈州立大学课程General Biochemistry BB 450/550:http://oregonstate.edu/instruct/bb450/450material/schedule450s17e.html
- csbsju课程Biochemistry Online:http://employees.csbsju.edu/hjakubowski/classes/ch331/protstructure/olunderstandconfo.html
- 蛋白质:http://refer.biovip.com/doc-view-334.html
肽键平面动画:
- https://www.sciencephoto.com/media/639617/view
- https://employees.csbsju.edu/hjakubowski/classes/ch331/protstructure/pp180to0.gif
- https://employees.csbsju.edu/hjakubowski/classes/ch331/protstructure/pp0to180.gif
- https://www.youtube.com/watch?v=Kewhg5spUjs
- https://www.youtube.com/watch?v=meNEUTn9Atg
{kind=link}
{kind=link}
蛋白质结构预测入门综述:
- 蛋白质二级结构预测方法的评价(唐一源,计算机与应用化学):http://www.cnki.com.cn/Article/CJFDTotal-JSYH200306003.htm
- 蛋白质结构预测(张阳,物理学报):http://www.cnki.com.cn/Article/CJFDTOTAL-WLXB201617012.htm
- 蛋白质三级结构预测算法综述(卜东波,计算机学报):http://www.cnki.com.cn/Article/CJFDTOTAL-JSJX201804002.htm
- 基于深度学习的八类蛋白质二级结构预测算法(杨伟,计算机应用):http://www.cnki.com.cn/Article/CJFDTOTAL-JSJY201705054.htm
- 蛋白质结构预测:梦想与现实(卜东波,信息技术快报):http://chinaxiv.org/user/download.htm?id=6478
蛋白质结构预测团队:
- DeepMind:https://deepmind.com/blog/alphafold/
- 密西根大学张阳团队:https://zhanglab.ccmb.med.umich.edu/
- 芝加哥大学许锦波团队:https://ttic.uchicago.edu/~jinbo/
- 哈佛医学院Mohammed AlQuraishi:https://scholar.harvard.edu/alquraishi
- 中科院计算所卜东波团队:http://bioinfo.ict.ac.cn/~dbu/
- 上海交大沈红斌团队:http://www.csbio.sjtu.edu.cn/
多谢博主的整理,让一个CSer快速知道蛋白结构预测大概是一个什么问题。我是西湖大学18级博士生,方向是做测序的,有机会多交流啊~
嗯嗯,我也是业余关注了一下蛋白质结构预测这个问题,感兴趣的话,可以再看看我整理的一篇Cell systems上使用深度学习预测蛋白质结构的文章:【论文速读】End-to-End Differentiable Learning of Protein Structure
您好,我是从知乎过来的(发了一条私信不小心删除了,没法发第二条了)。您的文章概念准确,思路清晰,把蛋白质结构预测问题分解成了若干个小问题。
很认同您的3个观点:1、有关“蛋白质结构预测”本身的最后一个问题是,为什么能仅仅通过一级结构的序列信息,预测得到其三级结构呢?2、综上所述,对于一条肽链,如果知道每个残基的三个扭转角ϕ、ψ和ω,则可以重构出肽链的主干部分的三维结构,这就像将极坐标转换为直角坐标一样容易。3、所以,每遇到一个新问题,都要自问一下,这个问题从原理上是否可解。
希望博主多多分享一些心得,互相学习交流。
您好,我是从知乎(发了私信不小心删除了,不能发第二条了)过来的。看了蛋白质结构预测这篇文章,思路清晰,层层递进,每个概念都解释的很清楚,而且思路严谨,可以说把这个问题阐述的很清楚了。
我很认同您说的3个观点:1、有关“蛋白质结构预测”本身的最后一个问题是,为什么能仅仅通过一级结构的序列信息,预测得到其三级结构呢?2、所以,每遇到一个新问题,都要自问一下,这个问题从原理上是否可解。3、综上所述,对于一条肽链,如果知道每个残基的三个扭转角ϕ、ψ和ω,则可以重构出肽链的主干部分的三维结构,这就像将极坐标转换为直角坐标一样容易。
希望您多多分享相关内容,以便学习交流。
另外不知道知乎还怎么能给您发私信呢?
您好,我是从知乎(发了私信不小心删除了,不能发第二条了)过来的。看了蛋白质结构预测这篇文章,思路清晰,层层递进,每个概念都解释的很清楚,而且思路严谨,可以说把这个问题阐述的很清楚了。
我很认同您说的3个观点:1、有关“蛋白质结构预测”本身的最后一个问题是,为什么能仅仅通过一级结构的序列信息,预测得到其三级结构呢?2、所以,每遇到一个新问题,都要自问一下,这个问题从原理上是否可解。3、综上所述,对于一条肽链,如果知道每个残基的三个扭转角ϕ、ψ和ω,则可以重构出肽链的主干部分的三维结构,这就像将极坐标转换为直角坐标一样容易。
希望您多多分享相关内容,以便学习交流。
另外不知道知乎还怎么能给您发私信呢?